
By Judy Carmody, Ph.D., Carmody Quality Solutions, LLC
Equipment qualification, including software validation for automated systems, is the process of demonstrating that equipment performs as intended. “As intended” is truly the key part of this process—defining the intended use of the equipment or system.
The process seems straightforward:
- Identify a business need for a piece of equipment or software.
- Gather a validation team and define and document the intended use and system requirements. The system requirements document (SRD), aka user requirements specification, forms the basis for all pending risk assessments and acceptance criteria. (A sample SRD template is available here.)
- Use the SRD to generate an RFP, or simply use the SRD as the specification to provide to potential vendors to source the needed equipment or software.
- Develop a project validation plan, including a traceability matrix, to outline the validation approach for the equipment and/or software.
- Create and implement the other qualification documents: installation qualification (IQ), operational qualification (OQ), and performance qualification (PQ), as necessary.
- Generate a validation summary report.
However straightforward this seems, typically this is not what happens. Usually, things go awry during the second step because we do not properly identify and document the intended use of the equipment/system, nor do we involve all of the potentially affected stakeholders in the decision-making process.
The typical scenario is:
- A business need is identified.
- The system owner identifies what is needed and contacts one or more vendors.
- Vendors are requested to provide proposals.
- Equipment/software is selected.
- Qualification/validation is conducted.
Upon first glance, it would appear this approach is acceptable; however, more often than not it isn’t, as seen in the following 483 observations:
- The FDA cited a testing laboratory for “failure to ensure laboratory instrumentation that is critical for assuring the quality of APIs is calibrated according to written procedures and at an established schedule.”
- The same lab was cited for not having any records indicating they had even qualified instrumentation prior to putting the “instrumentation into use.”
- An analytical laboratory was cited for providing its analysts with “PC administrator access that they utilized to manipulate raw data and test results.”
- A manufacturing facility was cited for not having “appropriate controls over computers or related systems to assure that changes in master production and control records or other records are instituted only by authorized personnel. Specifically, your firm’s manufacturing equipment are not 21 CFR part 11 compliant.”
- Another QC laboratory was cited for not having “appropriate controls over computers or related systems. There was a failure to maintain a backup file of data entered into the computer or related system.”
- A device company was cited for “failure to adequately ensure that all inspection, measuring, and test equipment, including mechanical, automated, or electronic inspection and test equipment, is suitable for its intended purposes and is capable of producing valid results according to established procedure, as required by 21 CFR 820.72(a). Specifically, your firm had no documentation that (b)(4) of testing equipment were qualified for their intended purpose per the associated standards.”
Such failures can be avoided by determining and documenting the intended use and needed requirements of the equipment and/or software. Getting this right is critical because all subsequent steps rely on properly executing this step.
Identify The Intended Use And System Requirements “Outside Of Your Own Bubble”
In essence, this translates to “How is the equipment/system going to be used?” and “What do you want it to do?” The answers to these questions should be determined before a vendor is contacted or a system is evaluated. More importantly, this exercise needs to take place with all of the stakeholders (i.e., the team) present. Not involving all stakeholders can yield negative, costly results.
Intended Use
To determine the intended use of the equipment/system, the team defines how or in what environment the equipment/system will be used. Will it be used in a regulated or research environment? Will it impact regulated systems? Defining this with members from different functional areas (e.g., facilities; IT; environmental, health, and safety; QA; validation; regulatory; manufacturing; clinical; laboratory) is critical to readily identify potential synergies and issues that can impact equipment/system selection.
What Quality Can Do
Quality can serve in a collaborative role, break down barriers, and bring people together to formalize processes and provide guidance, support, and education, as well as training to facilitate the execution of the process and appropriate SOPs and protocols. This ensures compliance to regulations and internal systems and that everyone is in tune with the equipment needed, its requirements, and how it will perform and operate.
System Requirements
Once the intended use is determined, we then answer the question, “What do you want the equipment/system to do?” To do this effectively, the team’s involvement is essential. This includes the team creating a list of all the equipment/system needs, including space requirements (e.g., it needs to fit here), procedural requirements (e.g., my test method or master batch record indicates this parameter is critical), functional requirements (e.g., it needs to operate in this manner), and regulatory requirements (e.g., it needs to allow for individual user sign-on with audit trails).
Once you come to a consensus on requirements, document everything in an SRD, as it is the basis for your RFP to vendors, as well as the other qualification documents (IQ, OQ, and PQ). After the system requirements are documented, categorize each requirement as mandatory, desired, critical, or non-critical. The mandatory/critical requirements must be tested as part of the qualification activities. The SRD connects to the IQ, OQ, and PQ and clearly identifies the testing of mandatory/critical system requirements outlined in each protocol.
The approach is no different than purchasing a television or a car. When you purchase anything, you create a list of needs for the intended purchase. When my husband and I needed a new car, he didn’t go off unilaterally and buy one. We discussed our requirements. Mine included regular gas (not premium), 4-wheel drive (thanks to New England winters), high fuel economy/MPG, four doors, and a sunroof. And since he services our cars, he added requirements: type of transmission, ease of service, cost of parts, and historical reliability. After discussing all options, we determined a Mazda 6 was suitable for our intended use (i.e., a reliable commuting car for me).
Imagine what would have happened if my husband had just brought home a car without any discussion because he liked the color or how much horsepower it had. Many companies make equipment/system purchasing decisions in this way.
Pay At The Beginning Or The End — You Decide
Does this approach involve a lot of work up-front? It can, but which is the better option: Doing the work up-front or having a $120,000 piece of equipment sitting on a palette in your laboratory unused because the intended use and system requirements were not properly defined and the equipment cannot do what is required? (I “inherited” such a piece of equipment after joining one company.)
People also get concerned about involving other departments because they may be hard to work with or are “cranky.” If you think your facilities or IT departments are cranky now, wait until you discover that the $120,000 piece of equipment also requires its own expensive HVAC system (not budgeted or planned for) or does not integrate with your existing network infrastructure and requires customized programming that takes months to implement.
Once the intended use and system requirements are documented in your SRD, you are in a much better position to develop proper qualification protocols (IQ, OQ, and PQ) and/or review vendor qualification protocols. The SRD serves as a means to identify the testing content for your validation/qualification protocols. Through the generation of a traceability matrix that maps the mandatory/critical requirements in your SRD to tests captured in the qualification protocols, you ensure all the important attributes of your equipment/system are qualified for their intended use.
Here is the entire process:
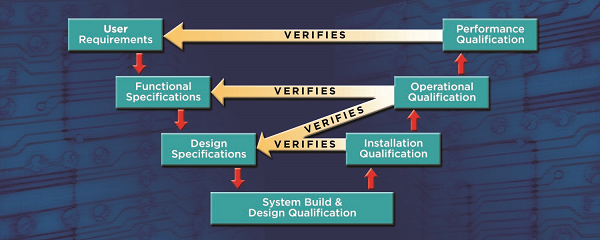
The final step in the process is the validation summary report which collates all of the validation activities tying the qualification results back to the intended use.
Final Thoughts
Using this approach for equipment qualification has a direct impact and improvement on data integrity. Why? Because, as the December 2018 FDA guidance Data Integrity and Compliance With Drug CGMP states, “If you validate the computer system but you do not validate it for its intended use, you cannot know if your workflow runs correctly.” The same applies to equipment, thereby calling into question any data generated on that piece of equipment. By taking the time to understand and document the intended use and equipment/system requirements, you ensure the equipment/system protocols are developed appropriately and, therefore, the equipment/system is qualified/validated correctly. When your process is well thought out and tested, you ensure a high level of integrity — that is, generated data that is reliable, accurate, and auditable.
These processes are less overwhelming when you collaborate and have all stakeholders involved as early as possible. Getting people around a table at the beginning can break down hurdles. It is easier for people to feel accountable and have a better, more invested understanding of the entire process from day one. They see the bigger picture when they are part of building the process and infrastructure. It will save time, money, and effort down the road.
For a sample system requirements document (SRD) template, click here.
About The Author:
 Judy Carmody, Ph.D., is the founder and principal consultant of Carmody Quality Solutions, LLC (CQS). She has more than 25 years of expertise in applied technology, bench chemistry, analytical development, validation, quality management, and senior leadership. She has built quality management systems for both startup and Fortune 500 companies.
Judy Carmody, Ph.D., is the founder and principal consultant of Carmody Quality Solutions, LLC (CQS). She has more than 25 years of expertise in applied technology, bench chemistry, analytical development, validation, quality management, and senior leadership. She has built quality management systems for both startup and Fortune 500 companies.
Carmody founded Avatar Pharmaceutical Services, an FDA-registered contract research organization that provided quality, submission-ready, customized analytical services in compliance with cGMP. She grew Avatar to more than 25 employees and more than 75 clients before it was purchased by a Boston-based pharmaceutical company in 2010. She holds a Ph.D. in analytical chemistry from Clark University in Worcester, Massachusetts. Please connect with her on LinkedIn.
